
Abstract
Purpose of review
To identify clues to disease activity and discuss therapy options.
Recent findings
The diagnostic evaluation includes documenting symmetrical proximal muscle damage by exam and MRI, as well as elevated muscle enzymes—aldolase, creatine phosphokinase, LDH, and SGOT—which often normalize with a longer duration of untreated disease. Ultrasound identifies persistent, occult muscle inflammation. The myositis-specific antibodies (MSA) and myositis-associated antibodies (MAA) are associated with specific disease course variations. Anti-NXP-2 is found in younger children and is associated with calcinosis; anti-TIF-1γ+ juvenile dermatomyositis has a longer disease course. The diagnostic rash—involving the eyelids, hands, knees, face, and upper chest—is the most persistent symptom and is associated with microvascular compromise, reflected by loss of nailfold (periungual) end row capillaries. This loss is associated with decreased bioavailability of oral prednisone; the bioavailability of other orally administered medications should also be considered. At diagnosis, at least 3 days of intravenous methyl prednisolone may help control the HLA-restricted and type 1/2 interferon–driven inflammatory process. The requirement for avoidance of ultraviolet light exposure mandates vitamin D supplementation.
Summary
This often chronic illness targets the cardiovascular system; mortality has decreased from 30 to 1–2% with corticosteroids. New serological biomarkers indicate occult inflammation: ↑CXCL-10 predicts a longer disease course. Some biologic therapies appear promising.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Juvenile dermatomyositis (JDM) is a rare disease with a characteristic rash and symmetrical proximal muscle weakness. Although it is classified as a vasculopathy [1], pharmacokinetic data suggests that the microvascular damage may not be limited to the skin [2] and muscle [3] and also may include the vasculature of the gastrointestinal tract, with a considerable negative impact on the bioavailability of some orally administered drugs [4]. The goal of this communication is to acquaint the reader with the common symptoms of this rare disease and provide current information concerning diagnosis and therapy [5]. Increasing clinical national and international collaboration [6] has facilitated the generation and analysis of larger datasets. There are more specific guidelines for diagnosis [7] and criteria for improvement [8], which are useful both for the western world and for the practitioners in China [9] and Japan [10]. This specific documentation has provided new clues concerning JDM pathophysiology, widening the spectrum of potential medical therapies.
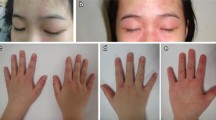
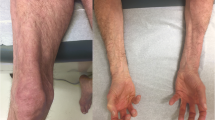
Juvenile dermatomyositis: symptoms and signs at diagnosis
The annual incidence of JDM by race in the USA is 3.4/million for white, non-Hispanic, 3.3/million for African Americans, and 2.7/million for Hispanic patients, with an overall girl-to-boy ratio of 2.3 girls:1 boy [11]. For the USA as a whole, the mean age at JDM diagnosis is 6.7 years for girls and 7.3 years for boys [12]. In our clinical population, which includes more than 500 children with definite/probable JDM [13, 14], 35.2% are under the age of 5 years at entry. The cutaneous features include Gottron’s sign—linear erythema–localized to the areas of the hands where the skin is stretched, over joints, while Gottron’s papules usually occur in areas of injury—fingers, elbows, and knees. Not only does the shawl sign occur on the upper chest, but also a typical rash may occur on areas of the body where there is pressure on the skin, such as the belt line. In addition to the classic rash and symmetrical proximal muscle weakness, initially identified by Bohan and Peter [13, 14], untreated children are shorter and lighter than their peers [15] and may develop the following complications: interstitial lung disease [16]; edema, either localized or general—secondary to capillary leak; and hair loss—secondary to scalp inflammation/edema. Cardiovascular damage includes impaired cardiac conduction [17, 18], and subsequent cardiac systolic dysfunction is predicted by cutaneous inflammation [19]. Evidence of microvascular damage can be documented by loss of capillary end row loops (ERL) in a child’s nailfold/periungual area [20, 21]. A well recognized physical finding—the presence of dilated capillaries at the eyelid margin—is often the last cutaneous sign to disappear [22]. Bone density is decreased in the untreated child with JDM and associated with an increased RANKL:osteoprotegerin ratio [23]; lipodystrophy is also a consequence of the JDM inflammatory process [24]. The onset of the characteristic rash, and the defining features of muscle damage with symmetrical proximal weakness, can run the gamut from insidious to very rapid progression.
The duration of untreated disease (DUD), the interval between the first symptom and the start of therapy, is a critical factor in understanding disease pathophysiology, impacting both the clinical findings and diagnostic laboratory data. Weaker children come to diagnosis earlier in their disease; those with only a mild rash—often much later [15]. The standard muscle enzymes are severely impacted as well (see serologic testing). As expected, gene expression profiles of muscle tissue are also modified by the DUD [25], which is noted infrequently in patient profiles.
Circumstances surrounding the onset of JDM
A variety of physical conditions may contribute to the development of JDM, including exposure to ultraviolet light [26]; the genetics of the patient appears to be associated with susceptibility to the increased dosage of UVB at equator-related latitudes [27]. Higher mean UV index exposure was associated with increased odds of using biologics or non-methotrexate disease-modifying anti-rheumatic drugs and skin ulceration, as well as developing calcinosis [28]. Increasing particulate pollution in the city atmosphere inflicts damage to the developing fetus and later in life [29]. Air pollution provokes oxidative stress, fostering the increasing development of a spectrum of autoimmune diseases [30], including JDM [29]. Exposure to an infectious agent may also play a role in precipitating the symptoms of JDM. A national study of 323 newly diagnosed children with JDM documented the presence of an antecedent illness, often in the upper respiratory tract, in the 3 months before JDM symptoms were noted [12].
Studies of seasonality of birth of children who later developed JDM [31] were associated with spring and fall seasonality of JDM onset in the Midwestern United States in the early 1980s. At the same time, there was an increase in both complement fixing and neutralizing antibody to Coxsackievirus B2 and B4 in newly diagnosed, untreated children with JDM [32]. Parvovirus 19 has also been implicated [33] as well as other agents. This apparent seasonality influence was lost, coincident with the increasing impact of global warming in about the mid-1980s, which stimulated a comprehensive re-evaluation of the impact of the accelerated rate of climate change [34]. An independent analysis of our gene expression profile data from muscles from children with untreated JDM recently concluded that the response to a double-stranded RNA agent [35] might be sufficient to initiate the interferon-based inflammatory progression typical of JDM [36].
The exact factors underlying the induction of the autoimmune response in JDM are not known, although multiple lines of evidence support the concept that an environmental exposure triggers disease in genetically susceptible individuals. Disease susceptibility has been mapped to the HLA locus on chromosome 6 using the genome-wide association methodology in a European population [37], [38]; C4A null alleles [39] and HLA-DRB1∗03:01 [38] are both risk factors for JDM. Differences in HLA associations with anti-TIF-1γ (p155/140) autoantibodies have been identified in adult-onset vs juvenile-onset DM [40], which may account for the differences in the disease course in these two otherwise similar populations. Increased CD4+ cells, TH-17, IL-6 [41], an expanded B cell population [42], and the emerging role of natural killer cells [43], as well as the PTPN22 R620W variant, are all linked to the increased risk of developing juvenile dermatomyositis.
Diagnostic testing overview
A wide spectrum of specific assessments has been compiled to aid the physician to evaluate skin and muscle involvement in both children and adults with inflammatory myopathies: JDM, juvenile polymyositis, DM, PM, and inclusion body myositis (IBM) [44]. A patient-reported outcomes system (PROMIS) has been developed for the age range of JDM disease onset—5–7 years of age—and found that a parent report cannot be substituted for the disclosure by the child [45]. A preliminary approach to evaluation of children with JDM was discussed using this PROMIS methodology [46].
Serologic and other blood assays
-
a)
Muscle enzymes: the standard diagnostic muscle enzymes (LDH, SGPT) normalize by 2.53 and 3.68 months respectively after symptom onset, while CPK and aldolase normalize by about 4.5 months [15]. Therefore, although CPK is a mainstay of the international classification criteria [7], it is not reliable after about 4 months between the first symptom and the clinical evaluation.
-
b)
Other “routine initial” testing: ANA (usually speckled), myositis-specific antibodies and myositis-associated antibodies, CBC, differential, CRP (elevated in overlap syndrome but not usually in JDM), ESR (usually normal range), U/A, BUN, Cr, neopterin—for macrophage activation; flow cytometry (for levels of both B cells and natural killer cells), C4 (for C4 null alleles), von Willebrand factor antigen (and blood group for normal ranges), vitamin D, DXA, and nailfold (periungual) capillary end row loop data [21].
-
c)
Myositis-specific antibodies (MSA) [47,48,49,50] (Table 1) vs myositis-associated antibodies (MAA) (Table 2) [48]: The importance of the expanding list of these antibodies cannot be overstated, for they provide a testable rationale for the heterogeneity of the inflammatory myopathies [51] in both adults and children [52, 53]. The most common type of MSA is antibody-TIF1-γ (transcriptional factor-1-γ) which is also called p155/140, occurring in 18–30% of children with JDM, and associated with a more prolonged and severe disease course [54], [55]. In over 25% of adults with inflammatory myopathy, the detection of both TIF1-γ and anti-NXP-2 (also called anti-MJ) is associated with the occurrence of a malignancy within a year of inflammatory muscle disease diagnosis [56]. Anti-NXP-2 has, as its target, NXP-2 and occurs in 15–22% of the children with JDM; it is associated with calcinosis, especially in children diagnosed below the age of 5 years [57] who manifest more severe muscle disease and gastrointestinal bleeding, resulting in a worse disease outcome with a lower functional status [58].
Overall, the next most common MSA is melanoma differentiation-associated gene 5 (MDA-5). In Japanese children, the frequency of MDA-5 is increased to 28% [59] vs 6% in the western world [52]. In China, the frequency of MDA-5 is similar both in children and in adults—30%—and associated with an amyopathic disease course in DM, interstitial lung disease (ILD), and digital ulcers [60]. A new biomarker, Krebs von den Lungen-6 (KL-6), appears to be useful in identifying children with ILD and is associated with increased IL-18 and ferritin [61]. ILD is increased in DM patients positive for MDA-5 who also may be clinically amyopathic (CADM) [51], as well as in children with inflammatory myopathy and other MSA, but can be reversed by aggressive therapy [62]. IV corticosteroids and cyclosporine A are frequently used for JDM with ILD; some biologics are promising.
The clinical picture is further complicated by the concurrent presence of myositis-associated antibodies (MAA), identifying overlap syndromes, such as those associated with antibodies to ribonuclear proteins (Table 2). Children positive for MAA follow a more chronic and relapsing disease course and have an increased frequency of calcinosis. An MAA, anti-Ro-52, is associated with ILD in children with JDM [63]; children positive for ILD also have increased levels of neopterin [16] which should prompt the use of high-resolution computerized tomography (CT) or ultrasound—to scan the lungs—as well as pulmonary function testing, if over the age of 6.
Diagnostic testing
Muscle strength and endurance
CMAS and MMT-8 are the mainstay of most clinical evaluations of muscle strength and endurance in children with JDM. To document the physical impairment consequent to the symmetrical proximal and truncal muscle weakness, it is very helpful to have an independent assessment by a physical therapist in a clinic at the time of a physician’s physical exam. However, the standardization of CMAS, to achieve a score of 52, was designed for healthy children aged 4–9 [64]. In contrast, for healthy children aged 4–5, we found that the achieved score was much lower, at 46, not 52 [65]. Over 26.3% of our newly diagnosed JDM are under age 4 at their first visit; interpreting CMAS data is hampered by lack of normal standards for younger ages.
Computerized tomography
Interstitial lung disease
This modality is used extensively in evaluating the presence and progression of ILD in JDM. Anti-Ro-52 antibody is associated with ILD in children with JDM [63] (see MSA/MAA above).
Volume of calcifications
A challenge in the treatment of JDM calcifications is to obtain convincing evidence that a medication is “working”—that the calcification volume is decreasing. Calcifications may respond slowly to immunosuppression or not at all. Single-slice CT as well as ultrasound (see below) can document the change in the volume of the calcifications as it grows or, in response to therapy, diminishes in size [66].
MRI: verification of target tissue involvement
Not only is MRI the method of choice for documenting inflammation and damage in all the proximal muscles at once, but it is also useful in evaluating unsuspected subcutaneous fat and fascial involvement [67]. When whole-body MRI (WB-MRI) utilizing short tau inversion recovery (STIR) was used to evaluate symptomatic and asymptomatic areas in muscle, subcutaneous tissue, and myofascial areas in 41 JDM patients, unsuspected areas of involvement were documented, and the pattern on MRI varied from patchy to diffuse [68]. This assessment is useful to identify persistent areas of inflammation, but in the young child with JDM, sedation is often needed, and the expense of the method is a deterrent.
Ultrasound in JDM
There is an emerging role for ultrasound in the evaluation of myositis in children with JDM. Clinical and laboratory parameters do not always parallel disease activity, so other methods of evaluation, such as muscle ultrasound, can provide complementary data in the evaluation of myositis [69]. Muscle thickness and echogenicity, which describes the ability of a tissue to reflect a sound wave, have been proposed as means of assessing disease activity in JDM [70]. In acute disease, muscle inflammation and edema are associated with an increase in muscle size and decrease in echogenicity, respectively, whereas in chronic disease, fatty infiltration and muscle atrophy are associated with decreased muscle size and increased echogenicity [70]. A significant correlation was found between echogenicity, CMAS, and serum CK [70]. Both fasciitis and calcinosis are also easily visualized, measured, and monitored by ultrasound [71], [72].
MRI is better suited to the initial JDM investigation given its ability to evaluate all the proximal muscles at once. Monitoring disease activity, on the other hand, may be more easily achieved with ultrasound. It is more readily available, transportable, cheaper, and non-invasive compared to MRI and can be performed as a point-of-care service in clinic. Changes in muscle thickness and muscle echogenicity are readily assessable by ultrasound. Ultrasound has a superior resolution and can detect more subtle changes in the muscles than MRI. In one study, which included seven children with JDM, ultrasound detected changes in the muscle fiber 1 year after whole-body MRI had normalized [69]. Thus, ultrasound may be a more sensitive measure of change and positive changes in the context of a negative MRI may be significant. For example, the presence of subclinical myositis may explain why some patients have weakness despite having a normal MRI and muscle enzymes [69].
Newer applications of ultrasound have explored vascularity within the muscles as a measure of disease activity and include the use of power Doppler (PDS), contrast-enhanced ultrasound (CEUS), and sonoelastography [73], [74]. Both power Doppler and color Doppler are used to evaluate blood flow and hyperemia within tissue. Color Doppler can also detect directional flow and can thus differentiate between arterial and venous circulation. In CEUS, microbubbles are infused intravenously and travel to areas of high perfusion, such as inflamed skeletal muscles. The degree of perfusion can be measured by CEUS and thereby serve as another marker of disease activity [75]. Sonoelastography takes advantage of the mechanical properties of tissues to assess inflammation, with stiffer tissue being more suggestive of inflammatory myositis [73]. Using these imaging modalities, the optimal site for biopsy can also be targeted, and ultrasound-guided muscle biopsy can be used to obtain the tissue. Current advances in adult myositis include developing machine learning methods to classify myositis by detecting changes in echogenicity of the affected tissue (muscle, fascia, subcutaneous tissue, fat, etc.) and correlating these results with different disease subtypes, for example, DM, PM, or IBM [76]. While computational approaches are emerging for classification purposes [77], machine learning in the context of ultrasonography has not been applied JDM, yet.
Increased multiple autoimmunity in JDM
Polyautoimmunity is not-uncommon in a person with a well-defined autoimmune disease [78]. The treating physician should consider screening for additional diagnoses in JDM patients with incipient symptoms, especially those treated with immunosuppressive medication. Immunosuppression may partially mask another emerging autoimmune disease, which occurs at higher than average rates in the families of patients with juvenile idiopathic arthritis, childhood lupus, and juvenile dermatomyositis [79]. In our study which evaluated familial autoimmunity within a three-generation pedigree of patients with JDM, SLE was reported in 50.1% of families [80]. A prospective analysis of our cohort of 590 JDM registry patients at the Ann & Robert H. Lurie Children’s Hospital of Chicago documented that the frequency of multiple autoimmunity is 8.7% (excluding diagnoses such as lupus or scleroderma that would be more representative of an underlying diagnosis of overlap syndrome). The additional autoimmune problem is most commonly vitiligo or psoriasis (n = 9 each), celiac disease (n = 8), and Hashimoto thyroiditis (n = 4). Four such patients have 2 additional autoimmune diagnoses, and one child with JDM has 5 additional diagnoses. Multiple autoimmunity has been described in smaller cohorts of JDM patients, with Hashimoto thyroiditis reported most frequently [78]. Multiple autoimmune diseases are a common feature of patients with diverse underlying syndromes of immune dysregulation including APECED (due to mutations in AIRE), CTLA-4 or LRBA haploinsufficiency, and Stat3 gain of function.
Accessible serum biomarkers
Many physicians are familiar with the phrase “clinically quiescent but immunologically active” which makes tapering of medications difficult. There is an urgent need for accessible serum biomarkers which can alert the physician to ongoing disease activity—despite the fact that the JDM appears to be “inactive”—both to guide therapy and to explore more effective interventions. A series of recent studies have emerged, ranging from a review of biomarkers of calcinosis [81] to studies of possible novel mechanisms propelling their formation [82]. A comprehensive series of investigations have identified serological indicators reflecting endothelial and vascular disruption [83,84,85]. For example, endoglin elevation and a low level of sICAM-1 were associated with loss of the end row loops (ERL) in the nailfold (periungual) area of untreated children with active JDM [85]. Loss of ERL is clearly associated with decreased bioavailability of orally administered prednisone compared to the same dose, given as an IV preparation [4]. Elevated levels of galectin 9, galectin 1, TNFR-2, and CXCL-10 indicted the need for intensification of treatment within the first 3 months, while high CXCL-10 at diagnosis also appeared to be predictive of a longer time to drug-free remission [85]. A recent study, using the Soma™Scan technology, identified serum proteins in JDM at various stages of therapy (untreated, on maximal therapy, and when being tapered) which are as follows: (a) responded to therapy, (b) rebounded when therapy was decreased, or (c) never responded to therapy at all—opening up new areas of investigation [86].
Treatment options
Although there is a fair amount of diversity in a physician’s selection of therapy [87], disease control can usually be obtained with currently available medications, which we hope has improved with the CARRA guidelines for care (see below). Of note, allogeneic bone marrow transplantation, sometimes used to treat inflammatory myopathy, can also elicit symptoms of inflammatory myopathy [88].
Physical therapy
The ventilatory capacity is often impaired in children with JDM because their truncal muscles are weakened. This muscle weakness contributes to low bone mass, aerobic deconditioning, and exercise intolerance. In contrast to the prolonged bed rest initially recommended for new-onset JDM, a randomized control trial of 26 JDM that variably assigned a 12-week home-based exercise program concluded that children who exercised had greater improvements than controls; but isometric strength and perception of fatigue were unaffected; no one required an increase in medications or hospitalization [89]. Exercise also helps reduce depression and assists in the recovery of muscle strength [90].
Medical therapy
Prior to corticosteroids, one third of the children diagnosed with inflammatory myopathy (JDM included) had calcinosis, one third died, and one third survived [91]. The scarcity of JDM patients led to the spontaneous use of a range of drugs. In contrast, a recent succession of useful protocols—which gauge the child’s response to therapy—has emerged from the Childhood Arthritis & Rheumatology Research Alliance (CARRA) group of pediatric rheumatologists. The initial guidelines for the treatment of moderately severe JDM [92] were followed by specific clinical treatment plans for JDM beyond the first 2 months [93], a protocol for the treatment of skin-predominant disease [94] as well as guidelines for the treatment of JDM with a persistent rash [95]. Of note, the actual usage of the medications to treat 320 children with JDM was recently reviewed [96].
Vitamin D
Inasmuch as dietary sources of vitamin D provide only 10% of the needed vitamin D and the recommended blood level for vitamin D for children is 30 IU or above, a daily supplementation with vitamin D of 1–2000 IU is recommended [97]. Low serum levels of vitamin D occur in people who have the idiopathic inflammatory myopathies (IIM) [98] including children with JDM [99]. Low levels are also found in subjects who are of increased age, are female, have a darker skin tone, and live in the northern latitude and stay there through the winter season of the year [97]. Since JDM often flare with sun/UVB exposure, they are usually sequestered during times of maximum exposure (see “Other therapies” below) and supplementation with exogenous vitamin D is required. Vitamin D deficiency is inversely associated with parathyroid hormone levels, which, as they rise, increases the risk of cardiovascular damage [100].
Corticosteroids
The mainstay of JDM medical therapy remains to be corticosteroids [101] which are life-saving, decreasing mortality to from 30 to 5% or much less [101, 102]. It is now customary to start with high-dose intravenous methyl prednisolone (maximal at 30 mg/kg), given in three or more consecutive daily doses once the diagnosis of JM is confirmed, as documented in a multinational PRINTO study [103]. Adverse reactions to IVMP in children are variable and encompass hyperglycemia, GI distress, hirsutism, breast enlargement in males, moon facies, thin skin with purple striae, osteoporosis, and CNS irritability, which includes an uncontrolled rage reaction [104]. Of note, absorption of oral prednisone is impaired in JDM patients whose nailfold end row capillary loops (ERL) fall below the normal range of 7 down to 5 ERL or less/3 mm [4]. This limitation was not considered in the recent recommendations by PRINTO for tapering of corticosteroids over time [105] and may vary with different medication preparations/conditions. Maintenance doses of corticosteroids range from 2 mg/kg/day to incrementally less. Gastric protection when corticosteroids are given is essential; options range from H2 blockers and proton pump inhibitors, to barrier protection such as Gaviscon®. The consequences of steroid therapy also include increased susceptibility to Pneumocystis jirovecii [106].
Hydroxychloroquine
A screening eye exam is required before starting this drug. The Academy of Ophthalmology Guidelines for testing for hydroxychloroquine retinopathy were changed from an “annual” exam to “5 years after starting the drug” [107]. The recommended dosage is 5 mg/kg/day; adverse reactions include abdominal pain ± nausea, which is the most common complaint, as well as cardiomyopathy, cardiac arrhythmia, elevated liver function tests, bone marrow suppression, and myopathy. Hydroxychloroquine is often not effective as a single drug [108]; it did not appear to improve the rash of 184 children with JDM [22]. The effect of hydroxychloroquine on the immune system includes increasing the lysosomal pH in antigen-presenting cells, resulting in decreased production of IL-1, IL-6, and TNF-α as well as blocking TLR7/9 on plasmacytoid dendritic cells, thus reducing the type 1 interferon signature which drives the inflammatory response [109]. In addition to its considerable anti-thrombotic effect [110], hydroxychloroquine is associated with a significant reduction in total cholesterol, triglycerides, and LDL levels [109] and is widely used for this purpose [111].
Methotrexate
The introduction of methotrexate after 1997 has allowed the use of less prednisone for disease control [96], [112]. The dosage is usually 1 mg/kg or 15 mg/m2 with a maximum of 25 mg/week, preferably given subcutaneously, accompanied by 1 mg/day of folic acid (on days other than the day that methotrexate is given) [113]. The most common adverse reaction is nausea and vomiting. Hair loss, which can be severe, and mouth sores, followed by bone marrow suppression, lung disease, lymphomas, and acute renal failure, are noted, but much less common [114]. Pregnancy is a contraindication; drug contact is associated with fetal malformations and miscarriage. High-risk patients should be screened for hepatitis B and C serology and if present, another drug should be used [114]. A recently identified mechanism of action is pro-respiratory and anti-growth by promoting AMPK signaling [115]. Withdrawal of this drug should be considered if the child is off steroids and in remission for at least a year.
Mycophenolate mofetil
Mycophenolate mofetil (MMF) and Myfortic should not be used interchangeably due to differences in concentration and absorption. Mycophenolate mofetil is given every 12 h at 20 mg/kg (maximal doses of 1000 mg/24 h). MMF is associated with congenital malformations and should be discontinued 7 weeks before a planned pregnancy. For girls, urine for pregnancy testing should be obtained at every visit when appropriate. This drug is a reversible, selective, and non-competitive inhibitor of inosine monophosphate, a critical enzyme in the de novo purine synthesis pathway required for lymphocyte proliferation [116]. At the moment, there are no guidelines to help identify who will benefit from MMF [117]. A study of 50 JDM patients documented that MMF both was steroid-sparing and decreased muscle and skin inflammation without a drop in the white count or an increased number of infections [118]. Adult DM with interstitial lung disease responded well to MMF. Although it is not widely recognized, vitamin D can lower the effective dose levels of MMF [119] which is a cause for concern: maintaining levels of vitamin D in a therapeutic range is critical for the well-being of children with JM, but unfortunately, a reliable testing for MMF levels is not widely available. Other adverse effects of MMF include diarrhea, bone marrow suppression, and reactivation of hepatitis B and C—children at high risk for hepatitis should be screened before the drug is given.
Tacrolimus
Much less used in recent years [120], [121], this agent has been effective, when combined with corticosteroids, in the treatment of a very difficult, relatively new form of inflammatory myopathy, defined by antibody to 3-hydroxy-3-methylglutaryl-coenzymeA reductase, and designated as HMGCR [122].
Intravenous immunoglobulin therapy
The half-life of intravenous immunoglobulin therapy (IVIG) is 3 weeks; the drug is usually given at 1–2 g/kg every month. One mode of action appears to be the induction of autophagy in peripheral blood mononuclear cells, thus reducing the production of inflammatory cytokines [123]. Hyaluronidase-facilitated immunoglobulin can be given subcutaneously at much higher doses [124, 125]. Tests for both immunoglobulin A (IgA) deficiency and myositis antibodies (MSA/MAA) should be obtained prior to the administration of the monomeric immunoglobulin, which is usually given intravenously. Reactions to IVIG, such as flushing, flu-like symptoms, headache, and fatigue, commonly occur about 24 h after the infusion and may last as long as 3 days, interfering with school attendance. Established guidelines suggest that IVIG should only be given to patients who have treatment failures or those who have become corticosteroid-dependent. The use of IVIG for interstitial lung disease was not recommended. There are a range of mechanisms proposed for the action of IVIG: cytokine and autoantibody neutralization, saturation of the Fc receptor, blocking receptor activation—which includes binding to anti-inflammatory receptors as well as other modes of cell modulation [124, 125]. Its use is advocated for JDM children who initially had both muscle and skin involvement but who continue to have rash as a persistent, but not predominant, complaint [95].
Cyclosporine A
This potent drug is a second-line agent and is given only when other, less toxic drugs have failed—in corticosteroid-resistant JM, often JDM with persistent rash [126]. A recent randomized trial of 139 JDM patients compared 3 treatment plans in 22 countries: after an induction period consisting of three daily doses of IV methylprednisolone at 30 mg/kg (1 g = maximal dose), the children were given either 2 years of oral prednisone alone vs prednisone + methotrexate vs prednisone + cyclosporine A (CyA). The study concluded that combined prednisone + either methotrexate or CyA performed better than prednisone alone, but more adverse reactions occurred with CyA (given at 4–5 mg/kg) requiring discontinuation of the drug [96]. In children with JDM, CyA can be used successfully at lower doses, 3 mg/kg, to maintain an 11th hour trough level of 80–110 ng/ml. An inhibitor of T cell activation, the pro-drug, CyA becomes activated after complexing with cyclophilin. This intracytoplasmic protein complex then inhibits calcineurin, a phosphatase that mediates the pharmacologic effects. Lipophilic in nature, a raised serum lipid level increases clearance of the drug, which therefore should be given before mealtime. Correspondingly, testing CyA levels should also performed before the child has eaten [55].
IV cyclophosphamide
This drug has been used successfully to treat refractory JDM, at a dosage of 500/m2 q 2 weeks ×3, then 750 mg/m2 q 3–4 weeks for 3–4 session [127]. Unfortunately, cyclophosphamide (CYC) induces oxidative stress and has a highly destructive effect on the reproductive organs. For ovaries, this can be averted by modern methods of ovary removal and storage with later re-implantation when therapy has been attenuated [128]. The cardiotoxic effects can also be muted by antioxidant supplementation [129].
Biologic agents: TNF inhibitors, rituximab, abatacept, and tocilizumab
Biologic agents are emerging as the targeted therapy for children with juvenile dermatomyositis [130] as well as other idiopathic inflammatory myopathies [131]. The possible immune pathways and their potentially specific drug targets in myositis have been recently reviewed [132].
TNF inhibitors
While these drugs are very useful in the treatment of other rheumatic diseases [133], this class of drugs is far from optimal in the treatment of inflammatory myopathy [134]. Another problem is that the use of TNF inhibitors to treat other rheumatic diseases, such as psoriatic arthritis, has precipitated the development of JDM [135], as well as inflammatory myopathies in adults [136, 137].
Rituximab
A monoclonal antibody directed against CD20, a surface marker for B cells, rituximab results in B cell depletion via several mechanisms which include complement fixation, antibody-dependent cellular cytotoxicity, and signaling of apoptosis. A close monitoring of the serum immunoglobulin G (IgG) levels is advisable, especially in the first year of therapy, for there is a 30–50% prevalence of hypogammaglobulinemia; some patients require an immunoglobulin replacement therapy [138]. Rituximab has been used successfully to treat JDM patients who failed first-line therapy [139, 140], [141]. A patient’s response to rituximab can be assessed by identifying the efficiency of bone marrow replacement of B cells. Forming new B cells requires B cell receptor recombination, which leads to the formation of KREC, a unique small piece of circular DNA that does not replicate as the B cells divide, so the KREC remains only in one cell and not the progeny (see Fig. 1). The site of recombination on the chromosomal DNA after the excision of KREC is known as the “joining code” (JC), which is found in all B cell progeny. The ratio of JC to KREC determined by qRT-PCR estimates with precision the number of B cell divisions that have occurred in the child’s B cell population. Therefore, it can provide a clue for the degree of peripheral B cell depletion [142]. The initial reports were followed by large multicenter randomized controlled clinical trial to evaluate the effectiveness of rituximab in both adult and pediatric patients with refractory myositis [139, 140], including cutaneous improvement [141]. Although the trial failed to show a significant difference between the study groups, post-study analysis suggested that specific MSA (anti-Jo-1 or anti-Mi-2) or young age (juvenile dermatomyositis vs adult myositis) predicted a better response to rituximab and that the most common side effects were infusion reactions, infections, and low blood counts [141]. We have found that those JDM patients who are treated with rituximab with complete or near complete B cell depletion will have a low JC-to-KREC ratio (see Fig. 1) which is associated with a more effective response to rituximab [143].

Cartoon of the kappa-deleting recombination excision circles (KREC) decreasing with cell division. The B cells originate in the bone marrow, located in this figure above the B cells. Khojah AM, Marin W, Morgan GA, Pachman LM. Kappa-deleting recombination excision circles (KREC) in B cells and serum B cell activating factor (BAFF): possible aids in predicting juvenile dermatomyositis response to rituximab. Arthritis Rheum [Internet]. 2018; 70(Suppl 10) abstract #11
Abatacept
This genetically engineered fusion protein is constructed from the FC portion of IgG1 and the extra-cellular domain of the cytotoxic T cell lymphocyte-associated protein 4 (CTLA-4). This drug decreases T cell activation by blocking the 2nd signal—CD80 and CD85—from antigen-presenting cells such as B cells [142]. Reports of adults with IIM who are not doing well on other treatments responded to abatacept and documented that the use of this drug along with methotrexate improved their outcome [118]. Used in conjunction with thiosulfate, abatacept was successful in eliciting regression of calcific lesions [144]. Of note, Dr. Rider at the NIH is still recruiting patients to complete their drug trial on abatacept in children with JDM and resistant calcinosis.
JAK kinase inhibitors
A dominant inflammatory pathway in JDM utilizes the type 1 interferons; their production is blocked by JAK kinase inhibitors. Tofacitinib blocks JAK 1 and 3, while baricitinib and ruxolitinib block JAK 1 and 2. They all are currently being tested for the therapeutic efficacy in refractory DM. Baricitinib was used successfully to treat a few children with JDM [145, 146], while tofacitinib was used to treat successfully 4 adult cases of DM [147], and ILD in some of the adults with MDA-5 [148]. Ruxolitinib, which affects circulating cytokine levels and regulates the activation of dendritic cells and T lymphocytes, improved the status of an adult with DM [149], and other dermatologic conditions responded as well [150]. The current recommendations are that women who are pregnant or breastfeeding should avoid this class of inhibitors [151].
Lenabasum
An oral synthetic cannabinoid receptor type 2 agonist, lenabasum decreased pulmonary flares in patients with cystic fibrosis. This was accomplished by inhibiting the polarization of macrophages into M1 and reducing their secretion of IL-8 and TNF-α [152]. Lenabasum has been used successfully to treat skin diseases in patients with diffuse cutaneous systemic sclerosis [153]. Pruritus is a common feature of both adult and juvenile dermatomyositis, and mast cells have been identified in both symptomatic and apparently normal sun-protected skin [154]. The “itch” has been linked to increased skin IL-31 [155]. Several recent phase 2 trials have indicated increased function and decreased skin symptoms with use of this emerging drug.
Other therapies
The treatment of cutaneous disease in JDM also includes avoidance of UVB exposure. The topical agents should contain UVA/B sunblock, such as titanium dioxide/oxide. A JDM child should avoid the sun during peak hours of 10–4 and wear photoprotective clothing—both purchased and home-rendered protective with the use of RIT SunGuardTM. Windows can be covered with a UV blocking film, and photosensitizing drugs, such as antibiotics (sulfa, tetracyclines), non-steroidal anti-inflammatory drugs, and diuretics should be avoided. The itch can be treated topically and orally with antihistamines, as well as by employing a systemic therapy for the inflammatory process of JDM.
Conclusion
The coordinated treatment of JDM is in its early stages, and there is great hope for more effective medications than those currently in use. The development of accessible serum biomarkers that could forecast disease flares would be very helpful and may aid in averting the premature cardiovascular compromise that is prevalent in adults who had JDM in childhood [156, 157].
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Papadopoulou C, McCann LJ. The vasculopathy of juvenile dermatomyositis. Front Pediatr. 2018;6:284.
Dugan EM, Huber AM, Miller FW, Rider LG. Photoessay of the cutaneous manifestations of the idiopathic inflammatory myopathies. DermatolOnlineJ. 2009;15(2):1.
Banker BQ, Victor M. Dermatomyositis (systemic angiopathy) of childhood. Medicine. 1966;45:261–89.
•• Rouster-Stevens KA, Gursahaney A, Ngai KL, Daru JA, Pachman LM. Pharmacokinetic study of oral prednisolone compared with intravenous methylprednisolone in patients with juvenile dermatomyositis. Arthritis Rheum. 2008;59(2):222–6. Documentation of decreased bioavaiability of oral prednisone vs IVMP in children with chronic JDM symptoms and loss of end row loops (ERL) in their nailfold capillaries.
Stringer E, Bohnsack J, Bowyer SL, Griffin TA, Huber AM, Lang B, et al. Treatment approaches to juvenile dermatomyositis (JDM) across North America: the Childhood Arthritis and Rheumatology Research Alliance (CARRA) JDM Treatment Survey. J Rheumatol. 2010;37(9):1953–61.
McCann LJ, Juggins AD, Maillard SM, Wedderburn LR, Davidson JE, Murray KJ, et al. The Juvenile Dermatomyositis National Registry and Repository (UK and Ireland)--clinical characteristics of children recruited within the first 5 yr. Rheumatology(Oxford). 2006;45(10):1255–60.
Lundberg IE, Tjärnlund A, Bottai M, Werth VP, Pilkington C, de Visser M, et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Arthritis Rheumatol. 2017;69(12):2271–82.
Rider LG, Aggarwal R, Pistorio A, Bayat N, Erman B, Feldman BM, et al. 2016 American College of Rheumatology/European League Against Rheumatism criteria for minimal, moderate, and major clinical response in juvenile dermatomyositis: an International Myositis Assessment and Clinical Studies Group/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Arthritis Rheumatol. 2017;69(5):911–23.
Zhang X, Yang X, Ji L, Zhang Z. Validation of 2017 classification criteria for adult and juvenile idiopathic inflammatory myopathies proposed by EULAR/ACR in Chinese patients. Int J Rheum Dis. 2019;22(7):1278–82.
Jinnin M, Ohta A, Ishihara S, Amano H, Atsumi T, Fujimoto M, et al. First external validation of sensitivity and specificity of the European League Against Rheumatism (EULAR)/American College of Rheumatology (ACR) classification criteria for idiopathic inflammatory myopathies with a Japanese cohort. Ann Rheum Dis. 2020;79(3):387–92.
Mendez EP, Lipton RB, Dyer A, Ramsey-Goldman R, Roftcher P, Bowyer S, et al. Incidence of juvenile dermatomyositis (JDM) 1995-98: results from the NIAMS Registry. Arthritis Care Res. 2003;49:300–5.
Pachman LM, Lipton R, Ramsey-Goldman R, Shamiyeh E, Abbott K, Mendez EP, et al. History of infection before the onset of juvenile dermatomyositis: results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Research Registry. Arthritis Rheum(Arthritis Care & Research). 2005;53(2):166–72.
Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). NEnglJ Med. 1975;292(7):344–7.
Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). NEnglJ Med. 1975;292(8):403–7.
Pachman LM, Abbott K, Sinacore JM, Amoruso L, Dyer A, Lipton R, et al. Duration of illness is an important variable for untreated children with juvenile dermatomyositis. JPediatr. 2006;148(2):247–53.
Prestridge A, Morgan G, Ferguson L, Huang CC, Pachman LM. Pulmonary function tests in idiopathic inflammatory myopathy: association with clinical parameters in children. Arthritis Care Res (Hoboken). 2013;65(9):1424–31.
Pachman LM, Cooke N. Juvenile dermatomyositis: a clinical and immunologic study. J Pediatr. 1980;96(2):226–34.
Ghosh R, Roy D, Dubey S, Abdelrahman K, Dey AK, Chatterjee S, et al. Juvenile dermatomyositis presenting as complete heart block in a 10-year-old girl. Paediatrics and international child health. 2020:1–4.
Schwartz T, Sanner H, Gjesdal O, Flatø B, Sjaastad I. In juvenile dermatomyositis, cardiac systolic dysfunction is present after long-term follow-up and is predicted by sustained early skin activity. Ann Rheum Dis. 2014;73(10):1805–10.
Smith RL, Sundberg J, Shamiyah E, Dyer A, Pachman LM. Skin involvement in juvenile dermatomyositis is associated with loss of end row nailfold capillary loops. JRheumatol. 2004;31(8):1644–9.
Christen-Zaech S, Seshadri R, Sundberg J, Paller AS, Pachman LM. Persistent association of nailfold capillaroscopy changes and skin involvement over thirty-six months with duration of untreated disease in patients with juvenile dermatomyositis. Arthritis Rheum. 2008;58(2):571–6.
Wang A, Morgan GA, Paller AS, Pachman LM. Skin disease is more recalcitrant than muscle disease: a long-term prospective study of 184 children with juvenile dermatomyositis. JAAD. 2020; in press.
Rouster-Stevens KA, Langman CB, Price HE, Seshadri R, Shore RM, Abbott K, et al. RANKL:osteoprotegerin ratio and bone mineral density in children with untreated juvenile dermatomyositis. Arthritis Rheum. 2007;56(3):977–83.
Khojah AMLV, Savani SI, Morgan GA, Huang C-C, Shore R, Bellm J, et al. Studies of 96 children with juvenile dermatomyositis: myositis specific antibody P155/140 (TIF-gamma) is associated with loss of nailfold capillaries, but not acquired lipodystrophy, determined by DXA. Arthritis Care Res (Hoboken). 2020. https://doi.org/10.1062/24535
Chen YW, Shi R, Geraci N, Shrestha S, Gordish-Dressman H, Pachman LM. Duration of chronic inflammation alters gene expression in muscle from untreated girls with juvenile dermatomyositis. BMCImmunol. 2008;9:43.
Okada S, Weatherhead E, Targoff IN, Wesley R, Miller FW. Global surface ultraviolet radiation intensity may modulate the clinical and immunologic expression of autoimmune muscle disease. Arthritis Rheum. 2003;48(8):2285–93.
• Parks CG, Wilkerson J, Rose KM, Faiq A, Noroozi Farhadi P, Long CS, et al. Association of ultraviolet radiation exposure with dermatomyositis in a national myositis patient registry. Arthritis Care Res (Hoboken). 2019; epublished 1–5. An uptodate summary of an evolving field.
Neely J, Long CS, Sturrock H, Kim S. Association of short-term ultraviolet radiation exposure and disease severity in juvenile dermatomyositis: results from the Childhood Arthritis and Rheumatology Research Alliance Legacy Registry. Arthritis Care Res (Hoboken). 2019;71(12):1600–5.
Vidotto JP, Pereira LA, Braga AL, Silva CA, Sallum AM, Campos LM, et al. Atmospheric pollution: influence on hospital admissions in paediatric rheumatic diseases. Lupus. 2012;21(5):526–33.
Zhao CN, Xu Z, Wu GC, Mao YM, Liu LN, Qian W, et al. Emerging role of air pollution in autoimmune diseases. Autoimmun Rev. 2019;18(6):607–14.
Vegosen L, Weinberg C, O'Hanlon TP, Miller F, Rider LG. Seasonal birth patterns of myositis suggest a role for early enviornmental exposures in etiology. Arthritis Rheum. 2007;56:2719–28.
Christensen ML, Pachman LM, Schneiderman R, Patel DC, Friedman JM. Prevalence of coxsackie B virus antibodies in patients with juvenile dermatomyositis. Arthritis Rheum. 1986;29:1365–70.
Mamyrova G, Rider LG, Haagenson L, Wong S, Brown KE. Parvovirus B19 and onset of juvenile dermatomyositis. Jama. 2005;294(17):2170–1.
Barnard RC, Morgan DL. The National Academy of Sciences offers a new framework for addressing global warming issues. Regulatory toxicology and pharmacology : RTP. 2000;31(1):112–6.
Musumeci G, Castrogiovanni P, Barbagallo I, Tibullo D, Sanfilippo C, Nunnari G, et al. Expression of the OAS gene family is highly modulated in subjects affected by juvenile dermatomyositis, resembling an immune response to a dsRNA virus infection. International journal of molecular sciences. 2018;19(9).
Tezak Z, Hoffman EP, Lutz JL, Fedczyna TO, Stephan DA, Krasnoselska I, et al. Expression profiling in DQA1*0501 children with juvenile dermatomyositis: a novel model of pathogenesis. J Immunol. 2002;168:4154–63.
Miller FW, Cooper RG, Vencovsky J, Rider LG, Danko K, Wedderburn LR, et al. Genome-wide association study of dermatomyositis reveals genetic overlap with other autoimmune disorders. Arthritis Rheum. 2013;65(12):3239–47.
Rothwell S, Cooper RG, Lundberg IE, Miller FW, Gregersen PK, Bowes J, et al. Dense genotyping of immune-related loci in idiopathic inflammatory myopathies confirms HLA alleles as the strongest genetic risk factor and suggests different genetic background for major clinical subgroups. Ann Rheum Dis. 2015;10.1136(20819):1–15.
Lintner KE, Patwardhan A, Rider LG, Abdul-Aziz R, Wu YL, Lundstrom E, et al. Gene copy-number variations (CNVs) of complement C4 and C4A deficiency in genetic risk and pathogenesis of juvenile dermatomyositis. Ann Rheum Dis. 2016;75(9):1599–606.
•• Rothwell S, Chinoy H, Lamb JA. Genetics of idiopathic inflammatory myopathies: insights into disease pathogenesis. Curr Opin Rheumatol. 2019;31(6):611–6. An excellent review of the specific HLA alleles associated with inflammatory myopathy.
Wilkinson MGL, Radziszewska A, Wincup C, Ioannou Y, Isenberg DA, Manson JJ, et al. Using peripheral blood immune signatures to stratify patients with adult and juvenile inflammatory myopathies. Rheumatology (Oxford). 2020;59(1):194–204.
Piper CJM, Wilkinson MGL, Deakin CT, Otto GW, Dowle S, Duurland CL, et al. CD19(+)CD24(hi)CD38(hi) B cells are expanded in juvenile dermatomyositis and exhibit a pro-inflammatory phenotype after activation through toll-like receptor 7 and interferon-alpha. Frontiers in immunology [Internet]. 2018 [PMC6024011]; 9:[1–15 pp.].
Throm AA, Alinger JB, Pingel JT, Daugherty AL, Pachman LM, French AR. Dysregulated NK cell PLCγ2 signaling and activity in juvenile dermatomyositis. JCI insight. 2018;3(22).
•• Rider LG, Werth VP, Huber AM, Alexanderson H, Rao AP, Ruperto N, et al. Measures of adult and juvenile dermatomyositis, polymyositis, and inclusion body myositis: Physician and Patient/Parent Global Activity, Manual Muscle Testing (MMT), Health Assessment Questionnaire (HAQ)/Childhood Health Assessment Questionnaire (C-HAQ), Childhood Myositis Assessment Scale (CMAS), Myositis Disease Activity Assessment Tool (MDAAT), Disease Activity Score (DAS), Short Form 36 (SF-36), Child Health Questionnaire (CHQ), Physician Global Damage, Myositis Damage Index (MDI), Quantitative Muscle Testing (QMT), Myositis Functional Index-2 (FI-2), Myositis Activities Profile (MAP), Inclusion Body Myositis Functional Rating Scale (IBMFRS), Cutaneous Dermatomyositis Disease Area and Severity Index (CDASI), Cutaneous Assessment Tool (CAT), Dermatomyositis Skin Severity Index (DSSI), Skindex, and Dermatology Life Quality Index (DLQI). Arthritis Care Res(Hoboken). 2011;2011:S118–S57. A very useful resource which compiles currently used measures.
Gao W, Yuan C, Zou Y, Lin H. Development and pilot testing a self-reported pediatric PROMIS app for young children aged 5-7 years. J Pediatr Nurs. 2020;53:74–83.
Patel REV, Lai J-S, Gray E, Chang R, Cella D, Ardalan K. Comparison of PROMIS computerized adaptive testing administered item banks vs fixed short forms in Juvenil myositis. Arthritis Rheum. 2019;71(10):abstract # 433.
Yasin SA, Schutz PW, Deakin CT, Sag E, Varsani H, Simou S, et al. Histological heterogeneity in a large clinical cohort of juvenile idiopathic inflammatory myopathy: analysis by myositis autoantibody and pathological features. Neuropathol Appl Neurobiol. 2019;45(5):495–512.
McHugh NJ, Tansley SL. Autoantibodies in myositis. Nat Rev Rheumatol. 2018;14(5):290–302.
Greenberg SA. Inclusion body myositis: clinical features and pathogenesis. Nat Rev Rheumatol. 2019;15(5):257–72.
Peterson LK, Jaskowski TD, La'ulu SL, Tebo AE. Antibodies to small ubiquitin-like modifier activating enzyme are associated with a diagnosis of dermatomyositis: results from an unselected cohort. Immunol Res. 2018;66(3):431–6.
Li D, Tansley SL. Juvenile dermatomyositis-clinical phenotypes. Curr Rheumatol Rep. 2019;21(12):74.
Tansley SL, Betteridge ZE, McHugh NJ. The diagnostic utility of autoantibodies in adult and juvenile myositis. Curr Opin Rheumatol. 2013;25(6):772–7.
Tansley SL, Snowball J, Pauling JD, Lissina A, Kuwana M, Rider LG, et al. The promise, perceptions, and pitfalls of immunoassays for autoantibody testing in myositis. Arthritis Res Ther. 2020;22(1):117.
Rider LG, Shah M, Mamyrova G, Huber AM, Rice MM, Targoff IN, et al. The myositis autoantibody phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine (Baltimore). 2013;92(4):223–43.
Pachman LM, Khojah AM. Advances in juvenile dermatomyositis: myositis specific antibodies aid in understanding disease heterogeneity. J Pediatr. 2018;195:16–27.
Fiorentino DF, Chung LS, Christopher-Stine L, Zaba L, Li S, Mammen AL, et al. Most patients with cancer-associated dermatomyositis have antibodies to nuclear matrix protein NXP-2 or transcription intermediary factor 1gamma. Arthritis Rheum. 2013;65(11):2954–62.
Tansley SL, Betteridge ZE, Shaddick G, Gunawardena H, Arnold K, Wedderburn LR, et al. Calcinosis in juvenile dermatomyositis is influenced by both anti-NXP2 autoantibody status and age at disease onset. Rheumatology (Oxford). 2014;53(12):2204–8.
Tansley SL, Simou S, Shaddick G, Betteridge ZE, Almeida B, Gunawardena H, et al. Autoantibodies in juvenile-onset myositis: their diagnostic value and associated clinical phenotype in a large UK cohort. J Autoimmun. 2017;10(1016).
Kobayashi I, Akioka S, Kobayashi N, Iwata N, Takezaki S, Nakaseko H, et al. Clinical practice guidance for juvenile dermatomyositis (JDM) 2018-Update. Mod Rheumatol. 2020;30(3):411–23.
So H, Ip RW, Wong VT, Yip RM. Analysis of anti-melanoma differentiation-associated gene 5 antibody in Hong Kong Chinese patients with idiopathic inflammatory myopathies: diagnostic utility and clinical correlations. Int J Rheum Dis. 2018;21(5):1076–81.
• Ye Y, Fu Q, Wang R, Guo Q, Bao C. Serum KL-6 level is a prognostic marker in patients with anti-MDA5 antibody-positive dermatomyositis associated with interstitial lung disease. J Clin Lab Anal. 2019;33(8):e22978. A relatively new test that may be helpful in the diagnosis of early lung disease in the rheumatic diseases.
Vega P, Ibarra M, Prestridge A, Pachman LM. Autoantibody to PL-12 (anti-Alanyl-tRNA synthetase) in an African American girl with juvenile dermatomyositis and resolution of interstitial lung disease. JRheumatol. 2011;38(2):394–5.
Sabbagh SP-FI, Kishi T, et al. Anti-Ro52 autoantibodies are associated with interstitial lung disease and more severe disease in patients with juvenile myositis. Ann Rheum Dis. 2019; Ann Rheum Dis. 2019;78:988–95. https://doi.org/10.1136/annrheumdis-2018-215004.
Rennebohm RM, Jones K, Huber AM, Ballinger SH, Bowyer SL, Feldman BM, et al. Normal scores for nine maneuvers of the Childhood Myositis Assessment Scale. Arthritis Rheum. 2004;51(3):365–70.
Quinones R, Morgan GA, Amoruso M, Field R, Huang CC, Pachman LM. Lack of achievement of a full score on the childhood myositis assessment scale by healthy four-year-olds and those recovering from juvenile dermatomyositis. Arthritis Care Res (Hoboken). 2013;65(10):1697–701.
Ibarra MRC, Morga GA, Sammet CL, Huang CC, Xu D, Targoff IN, et al. Monitoring change in volume of calcifications in juvenile myositis: a pilot study uusing low dose computed tomography (CT). Pediatric Rheumatology Online J. 2016;20:14.
Sakurai N, Hino-Shishikura A, Nozawa T, Kamide H, Ohara A, Nishimura K, et al. Clinical significance of subcutaneous fat and fascial involvement in juvenile dermatomyositis. Mod Rheumatol. 2019;29(5):808–13.
Malattia C, Damasio MB, Madeo A, Pistorio A, Providenti A, Pederzoli S, et al. Whole-body MRI in the assessment of disease activity in juvenile dermatomyositis. Ann Rheum Dis. 2014;73(6):1083–90.
Habers GE, Van Brussel M, Bhansing KJ, Hoppenreijs EP, Janssen AJ, Van Royen-Kerkhof A, et al. Quantitative muscle ultrasonography in the follow-up of juvenile dermatomyositis. Muscle Nerve. 2015;52(4):540–6.
Bhansing KJ, Hoppenreijs EP, Janssen AJ, van Royen-Kerkhof A, Nijhuis-Van der Sanden MW, van Riel PL, et al. Quantitative muscle ultrasound: a potential tool for assessment of disease activity in juvenile dermatomyositis. Scand J Rheumatol. 2014;43(4):339–41.
Bhansing KJ, Van Rosmalen MH, Van Engelen BG, Vonk MC, Van Riel PL, Pillen S. Increased fascial thickness of the deltoid muscle in dermatomyositis and polymyositis: an ultrasound study. Muscle Nerve. 2015;52(4):534–9.
• Orandi AB, Baszis KW, Dharnidharka VR, Huber AM, Hoeltzel MF. Assessment, classification and treatment of calcinosis as a complication of juvenile dermatomyositis: a survey of pediatric rheumatologists by the childhood arthritis and rheumatology research alliance (CARRA). Pediatr Rheumatol Online J. 2017;15(1):71. A recent summary of the status of a troubling complication of JDM.
Kubínová K, Dejthevaporn R, Mann H, Machado PM, Vencovský J. The role of imaging in evaluating patients with idiopathic inflammatory myopathies. Clin Exp Rheumatol. 2018;36(Suppl 114(5)):74–81.
Adler RSGG. Ultrasound in the evaluation of the inflammatory myopathies. Curr Rheumatology Rep. 2009;11:302–8.
Weber MA, Jappe U, Essig M, Krix M, Ittrich C, Huttner HB, et al. Contrast-enhanced ultrasound in dermatomyositis- and polymyositis. J Neurol. 2006;253(12):1625–32.
Burlina P, Billings S, Joshi N, Albayda J. Automated diagnosis of myositis from muscle ultrasound: exploring the use of machine learning and deep learning methods. PLoS One. 2017;12(8):e0184059.
Eng SWM, Olazagasti JM, Goldenberg A, Crowson CS, Oddis CV, Niewold TB, et al. A clinically and biologically based subclassification of the idiopathic inflammatory myopathies using machine learning. ACR open rheumatology. 2020;2(3):158–66.
Malagón C, Gomez MDP, Mosquera C, Vargas C, Gonzalez T, Arango C, et al. Juvenile polyautoimmunity in a rheumatology setting. Autoimmun Rev. 2019;18(4):369–81.
Prahalad S, McCracken CE, Ponder LA, Angeles-Han ST, Rouster Stevens KA, Vogler LB, et al. Familial autoimmunity in the Childhood Arthritis and Rheumatology Research Alliance registry. Pediatr Rheumatol Online J. 2016;14(1):14.
Niewold TB, Wu SC, Smith M, Morgan GA, Pachman LM. Familial aggregation of autoimmune disease in juvenile dermatomyositis. Pediatrics. 2011;127:1239–46.
Chung MP, Richardson C, Kirakossian D, Orandi AB, Saketkoo LA, Rider LG, et al. Calcinosis biomarkers in adult and juvenile dermatomyositis. Autoimmun Rev. 2020;19(6):102533.
•• Duvvuri B, Pachman LM, Morgan G, Khojah AM, Klein-Gitelman M, Curran ML, et al. Neutrophil extracellular traps in tissue and periphery in juvenile dermatomyositis. Arthritis Rheumatol. 2020;72(2):348–58. Positive identification of the role of PMNs –the NETS in JDM pathophysiology—
Wienke J, Deakin CT, Wedderburn LR, van Wijk F, van Royen-Kerkhof A. Systemic and tissue inflammation in juvenile dermatomyositis: from pathogenesis to the quest for monitoring tools. Front Immunol. 2018;9:2951.
Wienke J, Bellutti Enders F, Lim J, Mertens JS, van den Hoogen LL, Wijngaarde CA, et al. Galectin-9 and CXCL10 as biomarkers for disease activity in juvenile dermatomyositis: a longitudinal cohort study and multicohort validation. Arthritis Rheumatol. 2019;71(8):1377–90.
•• Wienke J, Pachman LM, Morgan GA, Yeo JG, Amoruso MC, Hans V, et al. Endothelial and inflammatory biomarker profile at diagnosis reflects clinical heterogeneity of juvenile dermatomyositis and is prognostic for response to treatment in two independent cohorts. Arthritis Rheumatol. 2020;72(7):1214–26. The identification of new biomarkers that may be predictive of outcome.
•• Tawalbeh SMMW, Morgan GA, Dang UJ, Hathout Y, Pachman LM. Novel serum protein biomarkers for juvenile dermatomyositis: a pilot study. 2019 Plos One. Novel serum protein biomarkers for juvenile dermatomyositis: a pilot study. PloS one. 2020; in press. The identification of new biomarkers that may be predictive of outcome.
Hinze CH, Speth F, Oommen PT, Haas JP. Current management of juvenile dermatomyositis in Germany and Austria: an online survey of pediatric rheumatologists and pediatric neurologists. Pediatr Rheumatol Online J. 2018;16(1):38.
Sakamoto K, Imamura T, Niwa F, Komori S, Ishihara Y, Shiga K, et al. Dermatomyositis developed in a recipient of allogeneic BMT; the differentiation of chronic GVHD and autoimmune disease. Bone Marrow Transplant. 2012;47(4):603–4.
Enders FB, Bader-Meunier B, Baildam E, Constantin T, Dolezalova P, Feldman BM, et al. Consensus-based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis. 2017;76(2):329–40.
Gualano B, Bonfa E, Pereira RMR, Silva CA. Physical activity for paediatric rheumatic diseases: standing up against old paradigms. Nat Rev Rheumatol. 2017;13(6):368–79.
Bitnum S, Daeschner C, Travis L, Dodge WF, Hopps H. Dermatomyositis. J Pediatr. 1964;64:101–31.
Huber AM, Giannini EH, Bowyer SL, Kim S, Lang B, Lindsley CB, et al. Protocols for the initial treatment of moderately severe juvenile dermatomyositis: results of a Children’s Arthritis and Rheumatology Research Alliance Consensus Conference. Arthritis Care Res(Hoboken). 2010;62(2):219–25.
Huber AM, Robinson AB, Reed AM, Abramson L, Bout-Tabaku S, Carrasco R, et al. Consensus treatments for moderate juvenile dermatomyositis: beyond the first two months. Arthritis Care Res(Hoboken). 2012;64(4):546–53.
Kim S, Kahn P, Robinson AB, Lang B, Shulman A, Oberle EJ, et al. Childhood Arthritis and Rheumatology Research Alliance consensus clinical treatment plans for juvenile dermatomyositis with skin predominant disease. Pediatr Rheumatol Online J. 2017;15(1):1.
Huber AM, Kim S, Reed AM, Carrasco R, Feldman BM, Hong SD, et al. Childhood Arthritis and Rheumatology Research Alliance Consensus clinical treatment plans for juvenile dermatomyositis with persistent skin rash. J Rheumatol. 2017;44(1):110–6.
Kishi T, Bayat N, Ward MM, Huber AM, Wu L, Mamyrova G, et al. Medications received by patients with juvenile dermatomyositis. Semin Arthritis Rheum. 2018;48(3):513–22.
Holick MF. The vitamin D deficiency pandemic: approaches for diagnosis, treatment and prevention. Rev Endocr Metab Disord. 2017;18(2):153–65.
Altieri B, Muscogiuri G, Barrea L, Mathieu C, Vallone CV, Mascitelli L, et al. Does vitamin D play a role in autoimmune endocrine disorders? A proof of concept. Rev Endocr Metab Disord. 2017;18(3):335–46.
Robinson AB, Thierry-Palmer M, Gibson KL, Rabinovich CE. Disease activity, proteinuria, and vitamin D status in children with systemic lupus erythematosus and juvenile dermatomyositis. J Pediatr. 2012;160(2):297–302.
Lee JH, O'Keefe JH, Bell D, Hensrud DD, Holick MF. Vitamin D deficiency an important, common, and easily treatable cardiovascular risk factor? J Am Coll Cardiol. 2008;52(24):1949–56.
Feldman BM, Rider LG, Reed AM, Pachman LM. Juvenile dermatomyositis and other idiopathic inflammatory myopathies of childhood. Lancet. 2008;371(9631):2201–12.
Hashkes PJ, Wright BM, Lauer MS, Worley SE, Tang AS, Roettcher PA, et al. Mortality outcomes in pediatric rheumatology in the US. Arthritis Rheum. 2010;62(2):599–608.
Ruperto N, Pistorio A, Oliveira S, Zulian F, Cuttica R, Ravelli A, et al. Prednisone versus prednisone plus ciclosporin versus prednisone plus methotrexate in new-onset juvenile dermatomyositis: a randomised trial. Lancet. 2016;387(10019):671–8.
Klein-Gitelman MS, Pachman LM. Intravenous corticosteroids: adverse reactions are more variable than expected in children. J Rheumatol. 1998;25(10):1995–2002.
Giancane G, Lavarello C, Pistorio A, Oliveira SK, Zulian F, Cuttica R, et al. The PRINTO evidence-based proposal for glucocorticoids tapering/discontinuation in new onset juvenile dermatomyositis patients. Pediatr Rheumatol Online J. 2019;17(1):24.
Ghembaza A, Vautier M, Cacoub P, Pourcher V, Saadoun D. Risk factors and prevention of Pneumocystis jirovecii pneumonia in patients with autoimmune and inflammatory diseases. Chest. 2020.
Marmor MF, Kellner U, Lai TY, Melles RB, Mieler WF. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy (2016 revision). Ophthalmology. 2016;123(6):1386–94.
Ang CC, Werth VP. Combination antimalerials in the treatment of cutaneous dermatomyositis. Arch Dermatolo. 2005;141:855–9.
Müller-Calleja N, Manukyan D, Canisius A, Strand D, Lackner KJ. Hydroxychloroquine inhibits proinflammatory signalling pathways by targeting endosomal NADPH oxidase. Ann Rheum Dis. 2017;76(5):891–7.
Carter AE, Eban R, Perrett RD. Prevention of postoperative deep venous thrombosis and pulmonary embolism. Br Med J. 1971;1(5744):312–4.
Rainsford KD, Parke AL, Clifford-Rashotte M, Kean WF. Therapy and pharmacological properties of hydroxychloroquine and chloroquine in treatment of systemic lupus erythematosus, rheumatoid arthritis and related diseases. Inflammopharmacology. 2015;23(5):231–69.
Harjacek J, Miller LC, Tucker LB, Schaller JB. Long-term follow-up for children receiving methotrexate for recalcitrant dermatomyositis. Arthritis Rheum. 1995;38(9, supplement):abstract #S231.
Al-Mayouf S, Al-Mazyed A, Bahabri S. Efficacy of early treatment of severe juvenile dermatomyositis with intravenous methylprednisolone and methotrexate. Clin Rheumatol. 2000;19(2):138–41.
Maksimovic V, Pavlovic-Popovic Z, Vukmirovic S, Cvejic J, Mooranian A, Al-Salami H, et al. Molecular mechanism of action and pharmacokinetic properties of methotrexate. Mol Biol Rep. 2020;47(6):4699–708.
Papadopoli DJ, Ma EH, Roy D, Russo M, Bridon G, Avizonis D, et al. Methotrexate elicits pro-respiratory and anti-growth effects by promoting AMPK signaling. Sci Rep. 2020;10(1):7838.
Villarroel MC, Hidalgo M, Jimeno A. Mycophenolate mofetil: an update. Drugs Today (Barc). 2009;45(7):521–32.
Mendoza-Pinto C, Pirone C, van der Windt DA, Parker B, Bruce IN. Can we identify who gets benefit or harm from mycophenolate mofetil in systemic lupus erythematosus? A systematic review. Semin Arthritis Rheum. 2017;47(1):65–78.
Rouster-Stevens KA, Morgan GA, Wang D, Pachman LM. Mycophenolate mofetil: a possible therapeutic agent for children with juvenile dermatomyositis. Arthritis Care Res (Hoboken). 2010;62(10):1446–51.
Wang X, Wang H, Shen B, Overholser BR, Cooper BR, Lu Y, et al. 1-Alpha, 25-dihydroxyvitamin D3 alters the pharmacokinetics of mycophenolic acid in renal transplant recipients by regulating two extrahepatic UDP-glucuronosyltransferases 1A8 and 1A10. Transl Res. 2016;178:54–62.e6.
Hassan J, van der Net JJ, van Royen-Kerkhof A. Treatment of refractory juvenile dermatomyositis with tacrolimus. Clin Rheumatol. 2008;27(11):1469–71.
Fasano S, Alves SC, Isenberg DA. Current pharmacological treatment of idiopathic inflammatory myopathies. Expert Rev Clin Pharmacol. 2016;9(4):547–58.
Feng F, Li Y, Ji S, Wang Q, Bu B. Tacrolimus combined with corticosteroids effectively improved the outcome of a cohort of patients with immune-mediated necrotising myopathy. Clin Exp Rheumatol. 2019;37(5):740–7.
Das M, Karnam A, Stephen-Victor E, Gilardin L, Bhatt B, Kumar Sharma V, et al. Intravenous immunoglobulin mediates anti-inflammatory effects in peripheral blood mononuclear cells by inducing autophagy. Cell Death Dis. 2020;11(1):50.
Angelotti F, Capecchi R, Giannini D, Mazzarella O, Rocchi V, Migliorini P. Long-term efficacy, safety, and tolerability of recombinant human hyaluronidase-facilitated subcutaneous infusion of immunoglobulin (Ig) (fSCIG; HyQvia((®))) in immunodeficiency diseases: real-life data from a monocentric experience. Clinical and experimental medicine. 2020. https://doi.org/10.1007/s10238-020-00633-4.
Chen Y, Wang C, Xu F, Ming F, Zhang H. Efficacy and tolerability of intravenous immunoglobulin and subcutaneous immunoglobulin in neurologic diseases. Clin Ther. 2019;41(10):2112–36.
Chighizola CB, Ong VH, Meroni PL. The use of cyclosporine a in rheumatology: a 2016 comprehensive review. Clin Rev Allergy Immunol. 2017;52(3):401–23.
Deakin CT, Campanilho-Marques R, Simou S, Moraitis E, Wedderburn LR, Pullenayegum E, et al. Efficacy and safety of cyclophosphamide treatment in severe juvenile dermatomyositis shown by marginal structural modeling. Arthritis & rheumatology (Hoboken, NJ). 2018;70(5):785–93.
Hussein RS, Khan Z, Zhao Y. Fertility preservation in women: indications and options for therapy. Mayo Clin Proc. 2020;95(4):770–83.
Ayza MA, Zewdie KA, Tesfaye BA, Wondafrash DZ, Berhe AH. The role of antioxidants in ameliorating cyclophosphamide-induced cardiotoxicity. Oxidative Med Cell Longev. 2020;2020:4965171.
Patwardhan A, Spencer CH. Biologics in refractory myositis: experience in juvenile vs. adult myositis; part II: emerging biologic and other therapies on the horizon. Pediatr Rheumatol Online J. 2019;17(1):56.
Khoo T, Limaye V. Biologic therapy in the idiopathic inflammatory myopathies. Rheumatol Int. 2020;40(2):191–205.
Oddis CV, Aggarwal R. Treatment in myositis. Nat Rev Rheumatol. 2018;14(5):279–89.
Angeles-Han ST, Ringold S, Beukelman T, Lovell D, Cuello CA, Becker ML, et al. 2019 American College of Rheumatology/Arthritis Foundation Guideline for the screening, monitoring, and treatment of juvenile idiopathic arthritis-associated uveitis. Arthritis Care Res (Hoboken). 2019;71(6):703–16.
Rouster-Stevens KA, Ferguson L, Morgan G, Huang CC, Pachman LM. Pilot study of etanercept in patients with refractory juvenile dermatomyositis. Arthritis Care Res (Hoboken). 2014;66(5):783–7.
Kim NN, Lio PA, Morgan GA, Jarvis JN, Pachman LM. Double trouble: therapeutic challenges in patients with both juvenile dermatomyositis and psoriasis. ArchDermatol. 2011;147(7):831–5.
Brunasso AM, Aberer W, Massone C. New onset of dermatomyositis/polymyositis during anti-TNF-α therapies: a systematic literature review. TheScientificWorldJournal. 2014;2014:179180.
Liu SW, Velez NF, Lam C, Femia A, Granter SR, Townsend HB, et al. Dermatomyositis induced by anti-tumor necrosis factor in a patient with juvenile idiopathic arthritis. JAMA dermatology. 2013;149(10):1204–8.
Khojah AM, Miller ML, Klein-Gitelman MS, Curran ML, Hans V, Pachman LM, et al. Rituximab-associated Hypogammaglobulinemia in pediatric patients with autoimmune diseases. Pediatr Rheumatol Online J. 2019;17(1):61.
Oddis CV, Reed AM, Aggarwal R, Rider LG, Ascherman DP, Levesque MC, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthritis Rheum. 2013;65(2):314–24.
Aggarwal R, Bandos A, Reed AM, Ascherman DP, Barohn RJ, Feldman BM, et al. Predictors of clinical improvement in rituximab-treated refractory adult and juvenile dermatomyositis and adult polymyositis. Arthritis Rheum. 2014;66(3):740–9.
Aggarwal R, Loganathan P, Koontz D, Qi Z, Reed AM, Oddis CV. Cutaneous improvement in refractory adult and juvenile dermatomyositis after treatment with rituximab. Rheumatology (Oxford). 2017;56(2):247–54.
Lorenzetti R, Janowska I, Smulski CR, Frede N, Henneberger N, Walter L, et al. Abatacept modulates CD80 and CD86 expression and memory formation in human B-cells. J Autoimmun. 2019;101:145–52.
Khojah AHV, Marin W, Morgan GA, Pachman LM. Kappa-deleting recombination excision circles (KREC) in B cells and serum B cell activating factor (BAFF): possible aids in predicting juvenile dermatomyositis response to rituximab. Arthritis Rheum [Internet]. 2018;70(Suppl 10):abstract #11.
Arabshahi B, Silverman RA, Jones OY, Rider LG. Abatacept and sodium thiosulfate for treatment of recalcitrant juvenile dermatomyositis complicated by ulceration and calcinosis. J Pediatr. 2012;160(3):520–2.
Papadopoulou C, Hong Y, Omoyinmi E, Brogan PA, Eleftheriou D. Janus kinase 1/2 inhibition with baricitinib in the treatment of juvenile dermatomyositis. Brain. 2019;142(3):e8.
Allenbach Y, Bolko L, Toquet S, Landon-Cardinal O, Benveniste O. Reply: Janus kinase 1/2 inhibition with baricitinib in the treatment of juvenile dermatomyositis. Brain. 2019;142(3):e9.
Moghadam-Kia S, Charlton D, Aggarwal R, Oddis CV. Management of refractory cutaneous dermatomyositis: potential role of Janus kinase inhibition with tofacitinib. Rheumatology (Oxford). 2019;58(6):1011–5.
Kurasawa K, Arai S, Namiki Y, Tanaka A, Takamura Y, Owada T, et al. Tofacitinib for refractory interstitial lung diseases in anti-melanoma differentiation-associated 5 gene antibody-positive dermatomyositis. Rheumatology (Oxford). 2018;57(12):2114–9.
Hornung T, Janzen V, Heidgen FJ, Wolf D, Bieber T, Wenzel J. Remission of recalcitrant dermatomyositis treated with ruxolitinib. N Engl J Med. 2014;371(26):2537–8.
Wu J, Smogorzewski J. Off-label studies on ruxolitinib in dermatology: a review. J of Dermatological Treatment. 2020;09:1–7.
You H, Xu D, Zhao J, Li J, Wang Q, Tian X, et al. JAK inhibitors: prospects in connective tissue diseases. Clin Rev Allergy Immunol. 2020. https://doi.org/10.1007/s12016-020-08786-6.
Tarique AA, Evron T, Zhang G, Tepper MA, Morshed MM, Andersen ISG, et al. Anti-inflammatory effects of lenabasum, a cannabinoid receptor type 2 agonist, on macrophages from cystic fibrosis. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2020. https://doi.org/10.1016/j.jcf.2020.03.015(5):1-7.
Hinchcliff M. Lenabasum for skin disease in patients with diffuse cutaneous systemic sclerosis. Arthritis Rheumatol. in press;2020(5).
Shrestha S, Wershil B, Sarwark J, Philipp T, Pachman L. Lesional and non-lesional skin from untreated juvenile dermatomyositis (JDM) displays increased mast cells and mature plasmacytoid dendritic cells. ArthritisRheum. 2010;69(0):2813–2822.
Kim HJ, Zeidi M, Bonciani D, Pena SM, Tiao J, Sahu S, et al. Itch in dermatomyositis: the role of increased skin interleukin-31. Br J Dermatol. 2018;179(3):669–78.
Eimer MJ, Brickman WJ, Seshadri R, Ramsey-Goldman R, McPherson DD, Smulevitz B, et al. Clinical status and cardiovascular risk profile of adults with a history of juvenile dermatomyositis. J Pediatr. 2011;159(5):795–801.
Silverberg JI, Kwa L, Kwa MC, Laumann AE, Ardalan K. Cardiovascular and cerebrovascular comorbidities of juvenile dermatomyositis in US children: an analysis of the National Inpatient Sample. Rheumatology (Oxford). 2018;57(4):694–702.
Acknowledgments
This compendium of observations and references could not have been acquired without the contributions of my clinical colleagues—Marisa Klein-Gitelman, MD, MPH; Michael Miller, MD; Brian E. Nolan, MD; Deidere de Ranieri, MD, RhMSUS; Kelly-Rouster-Stevens, MD; Maria Ibarra, MD; Megan Curran, MD; and many others, in addition to our dynamic research team—Wilfredo Marin, MS; Nahah Ahsan, BS, and especially—Maria Amoruso, MPH, CCRP; Gabrielle Morgan, MA, CCRP; and Amer Khojah, MD—as well as many, much appreciated, national and international collaborators. This work could not have been accomplished without the consistent financial help from The Cure-JM Foundation and the individual donors who have been unflagging in their support.
Funding
Supported by The Cure-JM Foundation; R21AR077565 NIH grant
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Lauren M. Pachman declares that she has no conflict of interest.
Brian E. Nolan declares that he has no conflict of interest.
Deidre De Ranieri declares that she has no conflict of interest.
Amer M. Khojah declares that he has no conflict of interest..
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Pediatric Rheumatology
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pachman, L.M., Nolan, B.E., DeRanieri, D. et al. Juvenile Dermatomyositis: New Clues to Diagnosis and Therapy. Curr Treat Options in Rheum 7, 39–62 (2021). https://doi.org/10.1007/s40674-020-00168-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40674-020-00168-5